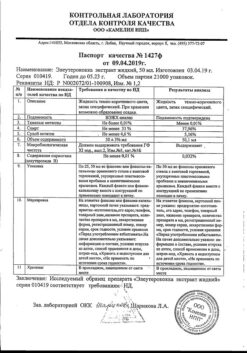
-
×
 Maalox, 15 ml suspension 30 pcs
2 × €20.92
Maalox, 15 ml suspension 30 pcs
2 × €20.92 -
×
Maalox, suspension 250 ml
1 × €11.00
Subtotal: €52.84
 Maalox, 15 ml suspension 30 pcs
2 × €20.92
Maalox, 15 ml suspension 30 pcs
2 × €20.92 Maalox, suspension 250 ml
1 × €11.00
Maalox, suspension 250 ml
1 × €11.00Subtotal: €52.84








€31.29
Liprimar is hypolipidemic, hypocholesterolemic.
Pharmacodynamics
Atorvastatin is a selective competitive inhibitor of HMG-CoA reductase, a key enzyme that converts 3-hydroxy-3-methylglutaryl-CoA to mevalonate, a precursor of steroids, including cholesterol, a synthetic hypolipidemic agent.
. In patients with homozygous and heterozygous familial hypercholesterolemia, nonfamilial forms of hypercholesterolemia and mixed dyslipidemia, atorvastatin reduces plasma levels of total cholesterol (CHs), chs-LDL and apo-B (apo-B), as well as chs-LBP and triglycerides (TG), causes unstable increase in HDL-C.
Atorvastatin reduces plasma cholesterol and lipoprotein concentrations by inhibiting HMG-CoA reductase and cholesterol synthesis in the liver and increasing the number of hepatic LDL receptors on the cell surface, which leads to increased capture and catabolism of hs-LDL.
Atorvastatin decreases formation of LDL-C and the number of LDL particles, causes a pronounced and sustained increase in LDL receptor activity in combination with favorable qualitative changes in LDL particles, and reduces LDL-C levels in patients with homozygous hereditary familial hypercholesterolemia resistant to therapy with other hypolipidemic agents.
Atorvastatin in doses from 10 to 80 mg reduces total cholesterol by 30-46%, LDL-C by 41-61%, apolipoprotein-B by 34-50% and TG by 14-33%. Results of therapy are similar in patients with heterozygous familial hypercholesterolemia, nonfamilial forms of hypercholesterolemia and mixed hyperlipidemia, including patients with insulin-independent diabetes.
In patients with isolated hypertriglyceridemia, atorvastatin decreases total cholesterol, chs-LDL, chs-LDLNP, apo-B and TG and increases chs-LDL.
In patients with dysbetalipoproteinemia, atorvastatin decreases intermediate-density lipoprotein cholesterol.
In patients with hyperlipoproteinemia of type IIa and IIb according to Frederickson mean increase of HDL-C content during treatment with atorvastatin (10-80 mg) in comparison with baseline is 5.1-8.7% and it is not dose dependent. There are significant dose-dependent reductions in the values of ratios: total cholesterol/Hs-LDL and hs-LDL/Hs-LDL by 29-44% and 37-55%, respectively.
Liprimar® in a dose of 80 mg significantly reduced the risk of coronary complications and mortality by 16% after 16-week course, and the risk of re-hospitalization for angina with signs of myocardial ischemia by 26%. In patients with different baseline concentrations of CHD Liprimar® causes a decrease in the risk of coronary complications and mortality (in patients with myocardial infarction without Q-wave and unstable angina, regardless of gender, age of the patient).
The decrease in plasma HDL-C correlates better with the drug dose than with its plasma concentration. The dose is adjusted according to the therapeutic effect (see section “Dosage and administration”).
Therapeutic effect is achieved 2 weeks after the start of therapy, reaches a maximum after 4 weeks and lasts for the duration of therapy.
Prevention of cardiovascular complications
. In the Anglo-Scandinavian Study of Cardiovascular Complications (lipid-lowering branch (ASCOT-LLA) the effect of atorvastatin on fatal and nonfatal outcomes of CHD was found that the effect of atorvastatin therapy at 10 mg dose was significantly greater than that of placebo, so it was decided to terminate the study early after 3.3 years instead of the intended 5 years.
Diabetes
The pooled study of the effect of atorvastatin on fatal and nonfatal cardiovascular outcomes in type 2 diabetes (CARDS) showed that atorvastatin therapy reduced the risk of the following cardiovascular complications regardless of gender, patient age or baseline CH-LD level.
Atherosclerosis
In a study of the reversal of coronary atherosclerosis with intensive lipid-lowering therapy (REVERSAL) with atorvastatin at a dose of 80 mg in patients with CHD, we found that the mean reduction in total atheroma volume (the primary efficacy criterion) since the start of the study was 0.4%.
Recurrent Stroke
. The Intensive Cholesterol Lowering Program (SPARCL) found that atorvastatin at a dose of 80 mg/day reduced the risk of recurrent fatal or nonfatal stroke in patients who had a stroke or transient ischemic attack (TIA) without a history of CHD by 15% compared with placebo. The risk of major cardiovascular complications and revascularization procedures was significantly reduced. Reduction of risk of cardiovascular disorders during atorvastatin therapy was observed in all groups except for the group including patients with primary or recurrent hemorrhagic stroke (7 in atorvastatin group versus 2 in placebo group).
Hemorrhagic stroke
The incidence of hemorrhagic or ischemic stroke (265 versus 311) or CHD (123 versus 204) was lower in patients treated with atorvastatin at a dose of 80 mg than in the control group.
Secondary prevention of cardiovascular complications
The New Target Trial (TNT) compared the effect of atorvastatin at doses of 80 and 10 mg/day on the risk of cardiovascular complications in patients with clinically confirmed CHD. Atorvastatin at a dose of 80 mg significantly reduced the development of the following complications.
Pharmacokinetics
Intake. Atorvastatin is rapidly absorbed after oral administration: Tmax in plasma is 1-2 hours. In women Cmax is 20% higher and AUC is 10% lower than in men. The degree of absorption and plasma concentration increase in proportion to the dose. Absolute bioavailability is about 14%, and systemic bioavailability of HMG-CoA reductase inhibitory activity is about 30%. Low systemic bioavailability is due to presystemic metabolism in the gastrointestinal mucosa and/or during first passage through the liver. Food slightly decreases rate and degree of drug absorption (by 25 and 9%, respectively, as evidenced by the results of Cmax and AUC determination, but decrease of low-density lipoprotein cholesterol (LDL-C) is similar to that during atorvastatin administration on an empty stomach. Despite the fact that after Atorvastatin administration in the evening its concentration in plasma is lower (Cmax and AUC, approximately by 30%) than after administration in the morning, decrease of HDL-C levels does not depend on the time of day, when the drug is taken.
Distribution. The average Vd of atorvastatin is about 381 l. The binding to plasma proteins is at least 98%. The ratio of content in erythrocytes/plasma is about 0.25, i.e. atorvastatin poorly penetrates into erythrocytes.
Metabolism. Atorvastatin is largely metabolized with the formation of ortho- and para-hydroxylated derivatives and various β-oxidation products. In vitro ortho- and para-hydroxylated metabolites have an inhibitory effect on HMG-CoA reductase, comparable with that of atorvastatin. Approximately 70% of the decrease of HMG-CoA reductase activity is due to the action of active circulating metabolites. Results of in vitro studies suggest that hepatic cytochrome CYP3A4 isoenzyme plays an important role in atorvastatin metabolism.
This fact is supported by increase of drug concentration in plasma when concomitant administration of erythromycin, which is an inhibitor of this isoenzyme. In vitro studies also showed that atorvastatin is a weak inhibitor of cytochrome CYP3A4 isoenzyme. Atorvastatin has no clinically significant effect on plasma concentrations of terfenadine, which is metabolized primarily with the participation of cytochrome CYP3A4 isoenzyme, therefore its significant effect on pharmacokinetics of other substrates of cytochrome CYP3A4 isoenzyme is unlikely (see section “Interaction”).
Elimination. Atorvastatin and its metabolites are excreted mainly with bile after hepatic and/or extrahepatic metabolism (atorvastatin does not undergo marked hepatic recirculation). T1/2 of preparation is approximately 14 h, hereby the inhibitory effect of the preparation with regard to HMG-CoA reductase is determined by activity of circulating metabolites approximately by 70% and lasts for about 20-30 h due to their presence. After oral administration, less than 2% of the administered dose of the drug is detected in the urine.
Particular groups of patients
Elderly patients. Atorvastatin concentrations in plasma of patients older than 65 years old are higher (Cmax – approximately by 40%, AUC – approximately by 30%) than those of adult patients of young age. No differences in efficacy and safety of the drug or achievement of hypolipidemic therapy goals were found in elderly patients compared to general population.
Children. No studies of pharmacokinetics of the drug in children have been conducted.
Inadequate renal function. Renal dysfunction does not influence atorvastatin concentration in plasma or its effect on lipid metabolism parameters, therefore there is no need to change the dose in patients with impaired renal function (see “Dosage and administration”).
Atorvastatin is not excreted during hemodialysis due to intense binding to plasma proteins.
Hepatic function insufficiency. Concentration of the drug is significantly increased (Cmax – approximately 16 times, AUC – approximately 11 times) in patients with alcoholic cirrhosis (stage B according to Child-Pugh classification).
Hypercholesterolemia:
– as an adjunct to diet to reduce elevated total cholesterol, LDL-C, apo-B and triglycerides in adults, adolescents and children 10 years of age or older with primary hypercholesterolemia, including familial hypercholesterolemia (heterozygous variant) or combined (mixed) hyperlipidemia (Fredrickson type IIa and IIb, respectively), when response to diet and other non-drug treatment methods are insufficient;
– to reduce elevated total cholesterol, LDL-C in adults with homozygous familial hypercholesterolemia as an adjunct to other lipid-lowering treatments (eg, LDL apheresis) or when such treatments are not available.
Prevention of cardiovascular diseases:
– prevention of cardiovascular events in adult patients at high risk of developing primary cardiovascular events, as an addition to the correction of other risk factors;
– secondary prevention of cardiovascular complications in patients with coronary artery disease in order to reduce mortality, myocardial infarction, stroke, re-hospitalization for angina pectoris and the need for revascularization.
Atorvastatin is a selective competitive inhibitor of HMG-CoA reductase, a key enzyme that converts 3-hydroxy-3-methylglutaryl-CoA to mevalonate, a precursor to steroids, including cholesterol. Synthetic lipid-lowering agent.
In patients with homozygous and heterozygous familial hypercholesterolemia, non-familial forms of hypercholesterolemia and mixed dyslipidemia, atorvastatin reduces the plasma concentrations of total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C) and apolipoprotein B (apo-B), as well as very low-density lipoprotein cholesterol (VLDL-C) and triglycerides (TG), causes an increase in the concentration of high-density lipoprotein cholesterol (HDL-C).
Atorvastatin reduces the concentration of cholesterol and LDL-C in the blood plasma by inhibiting HMG-CoA reductase and cholesterol synthesis in the liver and increasing the number of “liver” LDL receptors on the cell surface, which leads to increased uptake and catabolism of LDL-C.
Atorvastatin reduces the formation of LDL-C and the number of LDL particles, causes a pronounced and persistent increase in the activity of LDL receptors in combination with favorable qualitative changes in LDL particles, and also reduces the concentration of LDL-C in patients with homozygous hereditary familial hypercholesterolemia, resistant to therapy with other lipid-lowering drugs. Atorvastatin in doses from 10 mg to 80 mg reduces the concentration of cholesterol by 30-46%, LDL-C by 41-61%, apo-B by 34-50% and TG by 14-33%. The results of therapy are similar in patients with heterozygous familial hypercholesterolemia, non-familial forms of hypercholesterolemia and mixed hyperlipidemia, including patients with type 2 diabetes mellitus.
In patients with isolated hypertriglyceridemia, atorvastatin reduces the concentration of total cholesterol, LDL-C, VLDL-C, apo-B and TG and increases the concentration of HDL-C. In patients with dysbetalipoproteinemia, atorvastatin reduces the concentration of intermediate-density lipoprotein cholesterol (IDL-C).
In patients with hyperlipoproteinemia types IIa and IIb according to the Fredrickson classification, the average increase in HDL-C concentration during treatment with atorvastatin (10-80 mg) compared to baseline is 5.1-8.7% and is independent of dose. There is a significant dose-dependent decrease in the ratios: total cholesterol/HDL-C and LDL-C/HDL-C by 29-44% and 37-55%, respectively.
Atorvastatin at a dose of 80 mg significantly reduced the risk of ischemic complications and mortality by 16% after a 16-week course, and the risk of readmission for angina pectoris accompanied by signs of myocardial ischemia by 26% (myocardial ischemia reduction with intensive lipid-lowering therapy (MIRACL) study). In patients with different baseline LDL-C concentrations, atorvastatin causes a reduction in the risk of ischemic complications and mortality (in patients with non-Q wave myocardial infarction and unstable angina in men, women and in patients younger and older than 65 years).
The decrease in plasma LDL-C concentration correlates better with the dose of atorvastatin than with its plasma concentration. The dose is selected taking into account the therapeutic effect (see section “Method of administration and dosage”).
The therapeutic effect is achieved 2 weeks after the start of therapy, reaches a maximum after 4 weeks and persists throughout the entire period of therapy.
Prevention of cardiovascular complications
Atorvastatin at a dose of 10 mg reduces the relative risk of developing coronary complications (fatal coronary heart disease (CHD) and non-fatal myocardial infarction (MI) by 36%, total cardiovascular complications by 29%, fatal and non-fatal stroke by 26% (Atorvastatin Study in Patients with Hypertension and Risk Factors (ASCOT LLA)).
Diabetes mellitus
In patients with diabetes mellitus, atorvastatin therapy reduces the relative risk of major cardiovascular complications (fatal and nonfatal MI, silent myocardial ischemia, death due to exacerbation of coronary artery disease, unstable angina, coronary artery bypass grafting, percutaneous transluminal coronary angioplasty, revascularization procedures, stroke) by 37%, MI (fatal and non-fatal) by 42%, stroke (fatal and non-fatal) by 48%, regardless of gender, patient age or baseline LDL-C concentration (Atorvastatin in Type 2 Diabetes Mellitus Study (CARDS)).
Atherosclerosis
In patients with coronary artery disease, atorvastatin at a dose of 80 mg/day leads to a decrease in the total volume of atheroma by 0.4% over 1.8 months of therapy (reversal of coronary atherosclerosis during intensive lipid-lowering therapy study (REVERSAL)).
Repeated stroke
Atorvastatin 80 mg daily reduces the risk of recurrent fatal or nonfatal stroke in patients with a history of stroke or transient ischemic attack (TIA) without a history of CAD (Stroke Prevention with Intensive Cholesterol Lowering (SPARCL) Trial) by 16% compared with placebo. This significantly reduces the risk of major cardiovascular complications and revascularization procedures. A reduction in the risk of cardiovascular disorders during atorvastatin therapy was observed in all groups of patients, except for the one that included patients with primary or recurrent hemorrhagic stroke.
Secondary prevention of cardiovascular complications
In patients with coronary artery disease, atorvastatin at a dose of 80 mg, compared with 10 mg, significantly reduced the relative risk of major cardiovascular events by 22%, non-fatal myocardial infarction (not associated with revascularization procedures) by 22%, fatal and non-fatal stroke by 25% (comparison of high-intensity atorvastatin therapy and moderate-intensity therapy in patients with coronary artery disease (according to the study TNT)).
Suction
Atorvastatin is rapidly absorbed after oral administration: the time to reach its maximum concentration (TCmax) in blood plasma is 1-2 hours. In women, the maximum concentration of atorvastatin (Cmax) is 20% higher, and the area under the concentration-time curve (AUC) is 10% lower than in men. The degree of absorption and plasma concentration increase in proportion to the dose. The bioavailability of atorvastatin in tablet form is 95-99% compared to atorvastatin in solution form. Absolute bioavailability is about 14%, and systemic bioavailability of inhibitory activity against HMG-CoA reductase is about 30%. Low systemic bioavailability is due to first-pass metabolism in the mucous membrane of the gastrointestinal tract and/or during “first pass” through the liver. Eating slightly reduces the rate and extent of drug absorption (by 25% and 9%, respectively, as evidenced by the results of determining Cmax and AUC), however, the reduction in LDL-C is similar to that when taking atorvastatin on an empty stomach. Despite the fact that after taking atorvastatin in the evening, its plasma concentration is lower (Cmax and AUC by approximately 30%) than after taking it in the morning, the decrease in LDL-C concentration does not depend on the time of day at which the drug is taken.
Distribution
The mean volume of distribution of atorvastatin is approximately 381 L. Communication with blood plasma proteins is not less than 98%. The content ratio in red blood cells/blood plasma is about 0.25, i.e. Atorvastatin penetrates red blood cells poorly.
Metabolism
Atorvastatin is extensively metabolized to form ortho- and para-hydroxylated derivatives and various β-oxidation products. In vitro, ortho- and parahydroxylated metabolites have an inhibitory effect on HMG-CoA reductase comparable to that of atorvastatin. Approximately 70% of the decrease in HMG-CoA reductase activity occurs due to the action of active circulating metabolites. The results of in vitro studies suggest that the liver isoenzyme CYP3A4 plays an important role in the metabolism of atorvastatin. This fact is supported by an increase in the concentration of atorvastatin in the blood plasma while taking erythromycin, which is an inhibitor of this isoenzyme. In vitro studies have also shown that atorvastatin is a weak inhibitor of the CYP3A4 isoenzyme. Atorvastatin does not have a clinically significant effect on the plasma concentration of terfenadine, which is metabolized mainly by the CYP3A4 isoenzyme, so its significant effect on the pharmacokinetics of other CYP3A4 isoenzyme substrates is unlikely (see section “Interaction with other drugs”).
Removal
Atorvastatin and its metabolites are excreted mainly in bile after hepatic and/or extrahepatic metabolism (atorvastatin does not undergo significant enterohepatic recirculation). The half-life (T1/2) is about 14 hours, while the inhibitory effect of the drug on HMG-CoA reductase is approximately 70% determined by the activity of circulating metabolites and persists for about 20-30 hours due to their presence. After oral administration, less than 2% of the administered dose of the drug is found in the urine.
Atorvastatin is a substrate for the liver enzyme transporters OATP1B1 and OATP1B3. Metabolites of atorvastatin are substrates of OATP1B1.
Atorvastatin has also been identified as a substrate of the efflux transporters MDR1 and breast cancer resistance protein, which may limit the intestinal absorption and biliary clearance of atorvastatin.
Special patient groups
Elderly patients
Plasma concentrations of atorvastatin in patients over 65 years of age are higher (Cmax approximately 40%, AUC approximately 30%) than in younger adult patients. There were no differences in the effectiveness and safety of the drug, or the achievement of lipid-lowering therapy goals in elderly patients compared with the general population.
Children
In an 8-week open-label study, children (aged 6-17 years) with heterozygous familial hypercholesterolemia and baseline LDL-C concentrations >= 4
mmol/L received atorvastatin therapy in the form of 5 mg or 10 mg chewable tablets or 10 mg or 20 mg film-coated tablets once daily, respectively. The only significant covariate in the pharmacokinetic model of the atorvastatin population was body weight. The apparent clearance of atorvastatin in children did not differ from that in adult patients when allometrically measured by body weight. In the range of action of atorvastatin and o-hydroxyatorvastatin, a consistent decrease in LDL-C and cholesterol was observed.
Gender
Plasma concentrations of atorvastatin in women differ from those in men (approximately 20% higher for Cmax and 10% lower for AUC). However, no clinically significant differences in the effect on lipids were observed between men and women.
Kidney failure
Impaired renal function does not affect the concentration of atorvastatin in the blood plasma or its effect on lipid metabolism, and therefore no dose change is required in patients with impaired renal function (see section “Dosage and Administration”).
There have been no studies of atorvastatin in patients with end-stage renal disease. Atorvastatin is not excreted during hemodialysis due to intense binding to plasma proteins.
Liver failure
The concentration of the drug is significantly increased (Cmax approximately 16 times, AUC approximately 11 times) in patients with alcoholic cirrhosis (Child-Pugh class B) (see section “Contraindications”).
Polymorphism SLCO1B1
Hepatic uptake of all HMG-CoA reductase inhibitors, including atorvastatin, occurs via the OATP1B1 transporter. Patients with the SLCO1B1 genetic polymorphism are at risk of increased atorvastatin exposure, which may lead to an increased risk of rhabdomyolysis. A polymorphism in the gene encoding OATP1B1 (SLCO1B1 c.521CC) is associated with a 2.4-fold increase in atorvastatin exposure (AUC) compared to patients without such a genotypic change (c.521TT). Impaired hepatic uptake of atorvastatin due to genetic disorders may also occur in these patients. Possible effects on effectiveness are unknown.
Effect on the liver
As with the use of other lipid-lowering drugs of this class, when using the drug Liprimar®, a moderate increase (more than 3 times compared with the upper limit of normal) in the activity of “liver” transaminases AST and ALT was noted. A persistent increase in serum activity of “liver” transaminases (more than 3 times compared to the upper limit of normal) was observed in 0.7% of patients receiving Liprimar®. The incidence of such changes when using the drug in doses of 10 mg, 20 mg, 40 mg and 80 mg was 0.2%, 0.2%, 0.6% and 2.3%, respectively. Increased activity of liver transaminases was usually not accompanied by jaundice or other clinical manifestations. When the dose of Liprimar® was reduced, or the drug was temporarily or completely discontinued, the activity of “liver” transaminases returned to the original level. Most patients continued taking Liprimar® at a reduced dose without any clinical consequences.
Before starting therapy, 6 weeks and 12 weeks after starting the use of Liprimar® or after increasing its dose, it is necessary to monitor indicators
liver functions. Liver function should also be monitored when clinical signs of liver damage appear. In case of increased activity of “liver” transaminases, ALT and AST should be monitored until it returns to normal. If an increase in AST or ALT activity by more than 3 times compared to the upper limit of normal persists, it is recommended to reduce the dose or discontinue the drug Liprimar® (see section “Side effects”).
Liprimar® should be used with caution in patients who consume significant amounts of alcohol and/or have a history of liver disease. Active liver disease or persistently elevated activity of hepatic transaminases of unknown origin are a contraindication to the use of Liprimar® (see section “Contraindications”).
Effect on skeletal muscles
Myalgia was observed in patients receiving Liprimar® (see section “Side effects”). The diagnosis of myopathy should be considered in patients with diffuse myalgia, muscle soreness or weakness, and/or a marked increase in CPK activity (more than 10 times the upper limit of normal). Therapy with Liprimar® should be discontinued in the event of a marked increase in CPK activity, in the presence of confirmed myopathy or suspicion of its development. The risk of myopathy increased with simultaneous use of drugs that increase the systemic concentration of atorvastatin (see sections “Interaction with other drugs” and “Pharmacokinetics”). Many of these drugs inhibit CYP3A4-mediated metabolism and/or drug transport. It is known that the CYP3A4 isoenzyme is the main liver isoenzyme involved in the biotransformation of atorvastatin. When using Liprimar® in combination with fibrates, erythromycin, immunosuppressants, azole antifungals, HIV/HCV protease inhibitors, hepatitis C viral non-structural protein inhibitors (NS5A/NS5B), letermovir or nicotinic acid in lipid-lowering doses (more than 1 g/day), the doctor must carefully weigh the expected benefits of treatment and the possible risks. Patients should be regularly monitored for muscle pain or weakness, especially during the first months of therapy and during dosage increases of any of these agents. If combination therapy is necessary, the possibility of using lower initial and maintenance doses of the above drugs should be considered (see section “Dosage and Administration”). The simultaneous use of atorvastatin and fusidic acid is not recommended, therefore, temporary withdrawal of atorvastatin is recommended during treatment with fusidic acid. In such situations, periodic monitoring of CPK activity can be recommended, although such monitoring does not prevent the development of severe myopathy (see section “Interaction with other drugs”).
There have been very rare reports of the development of immune-mediated necrotizing myopathy (IONM) during or after treatment with certain statins (see section “Side effects”). IONM is clinically characterized by persistent proximal muscle weakness and elevated serum creatine kinase levels that persist despite discontinuation of statin treatment, the presence of antibodies to HMG CoA reductase, and improvement with immunosuppressive medications.
Before treatment
Atorvastatin should be prescribed with caution to patients with factors predisposing to the development of rhabdomyolysis. Monitoring of CPK activity should be carried out in the following cases before starting atorvastatin therapy:
– renal dysfunction,
– hypothyroidism,
– the patient has a history or family history of hereditary muscle disorders,
– already suffered toxic effects of HMG-CoA reductase inhibitors (statins) or fibrates on muscle tissue,
– history of liver disease and/or patients drinking alcohol in significant quantities,
– in patients over the age of 70 years, the need for CK control should be assessed, given that these patients already have factors predisposing to the development of rhabdomyolysis,
– situations in which an increase in the concentration of atorvastatin in the blood plasma is expected, such as interactions with other drugs (see section “Interaction with other drugs”).
In such situations, the risk/benefit ratio should be assessed and medical monitoring of the patient’s condition should be carried out.
In case of a significant increase in CPK activity (more than 5 times the upper limit of normal), atorvastatin therapy should not be started.
When using the drug Liprimar®, as well as other HMG-CoA reductase inhibitors, rare cases of rhabdomyolysis with acute renal failure caused by myoglobinuria have been described. A risk factor for the development of rhabdomyolysis may be pre-existing renal impairment. Such patients should be provided with more careful monitoring of the musculoskeletal system. If symptoms of possible myopathy appear or there are risk factors for the development of renal failure due to rhabdomyolysis (for example, severe acute infection, hypotension, major surgery, trauma, metabolic, endocrine and fluid-electrolyte disturbances, uncontrolled seizures), therapy with Liprimar® should be temporarily stopped or completely discontinued.
Attention! Patients should be advised to seek immediate medical attention if they experience unexplained pain or muscle weakness, especially if accompanied by malaise or fever.
Hemorrhagic stroke
After a special analysis of a clinical trial involving 4,731 patients without coronary artery disease who had a stroke or transient ischemic attack (TIA) within the previous 6 months who were prescribed atorvastatin 80 mg/day, a higher incidence of hemorrhagic stroke was found in the atorvastatin 80 mg group compared with the placebo group (55 in the atorvastatin group vs. 33 in the placebo group). Patients with hemorrhagic stroke at study entry had a higher risk of recurrent hemorrhagic stroke (7 in the atorvastatin group versus 2 in the placebo group). However, patients receiving atorvastatin 80 mg/day had fewer strokes of any type (265 vs. 311) and fewer cardiovascular events (123 vs. 204) (see Pharmacodynamics section).
Diabetes mellitus
Some evidence suggests that HMG-CoA reductase inhibitors (statins), as a class, may lead to elevated plasma glucose concentrations, and some patients at high risk for developing diabetes mellitus may develop a hyperglycemic state requiring treatment as in diabetes mellitus. However, this risk does not outweigh the benefits of therapy with HMG-CoA reductase inhibitors (statins) in terms of vascular risks, so this cannot be a reason to discontinue therapy. Patients at risk (fasting blood glucose concentration from 5.6 to 6.9 mmol/l, BMI > 30 kg/m2, elevated plasma triglyceride concentrations, arterial hypertension) should be under medical supervision, including monitoring of blood biochemical parameters, in accordance with local recommendations.
Interstitial lung disease
During therapy with certain HMG-CoA reductase inhibitors (statins), especially during long-term therapy, isolated cases of interstitial lung disease have been reported. Shortness of breath, nonproductive cough, and deterioration in general health (fatigue, weight loss, and fever) may occur. If interstitial lung disease is suspected in a patient, atorvastatin therapy should be discontinued.
Endocrine function
When using HMG-CoA reductase inhibitors (statins), including atorvastatin, there have been cases of increased glycosylated hemoglobin (HbA1) and fasting plasma glucose concentrations. However, the risk of hyperglycemia is lower than the degree of reduction in the risk of vascular complications while taking HMG-CoA reductase inhibitors (statins).
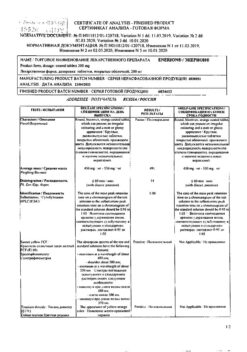
Atorvastatin
1 film-coated tablet contains:
Active ingredient: atorvastatin calcium (equivalent to 20 mg atorvastatin)
Excipients: calcium carbonate, microcrystalline cellulose (PH-101), lactose monohydrate, croscarmellose sodium, polysorbate-80, hyprolose (hydroxypropylcellulose), magnesium stearate;
film coating: opadry white YS-1-7040 (contains hypromellose (hydroxypropyl methylcellulose), macrogol (polyethylene glycol 8000), titanium dioxide, talc), simethicone emulsion (contains simethicone, polysorbate 65, methylcellulose, PEG-8 stearate, glycerol stearate, xanthan gum, sorbic acid, benzoic acid, sulfuric acid, water), herbal wax
Women of childbearing age
Women of childbearing age should use appropriate methods of contraception during treatment (see section “Contraindications”).
Pregnancy
Liprimar® is contraindicated during pregnancy (see section “Contraindications”). Safety of use during pregnancy has not been confirmed. Controlled clinical studies with atorvastatin have not been conducted among pregnant women. Rare cases of congenital anomalies have been reported after fetal exposure to HMG-CoA reductase inhibitors (statins) in utero. Animal studies have shown reproductive toxicity.
When a pregnant woman takes atorvastatin, the levels of mevalonate, which is a precursor for cholesterol biosynthesis, may decrease in the fetus. Atherosclerosis is a chronic process and, in general, discontinuation of lipid-lowering drugs during pregnancy has only a minor effect on the long-term risk associated with primary hypercholesterolemia.
In this regard, Liprimar® should not be prescribed to pregnant women, women planning pregnancy, or if pregnancy is suspected. It is necessary to discontinue taking Liprimar® during pregnancy or until the absence of pregnancy is established (see section “Contraindications”).
Breastfeeding period
It is not known whether atorvastatin or its metabolites are excreted into breast milk in humans.
In studies in rats, plasma concentrations of atorvastatin and its active metabolites were similar to those in milk. Due to the risk of serious side effects, women taking Liprimar® should not breastfeed their children (see section “Contraindications”). The use of Liprimar® is contraindicated during breastfeeding (see section “Contraindications”).
Fertility
In animal studies, atorvastatin had no effect on fertility in males or females.
Hypersensitivity to any component of the drug.
Active liver disease or an increase in the activity of “liver” transaminases in the blood plasma of unknown origin by more than 3 times compared with the upper limit of normal. Pregnancy.
Breastfeeding period.
Women of childbearing age who are not using adequate contraceptive methods. Age up to 18 years (there is insufficient clinical data on the effectiveness and safety of the drug in this age group), with the exception of heterozygous familial hypercholesterolemia (use is contraindicated in children under 10 years of age). Concomitant use with fusidic acid.
Congenital lactase deficiency, lactose intolerance, glucose-galactose malabsorption.
Liprimar® is generally well tolerated; Adverse reactions are usually mild and transient.
Adverse reactions are distributed according to frequency according to the following classification: often – ≥ 1/100 to < 1/10, uncommon - ≥ 1/1000 to < 1/100, rarely - ≥ 1/10000 to < 1/1000, very rare - < 1/10000, unknown - cannot be estimated based on available data.
Infectious and parasitic diseases:
Often – nasopharyngitis.
Blood and lymphatic system disorders:
Rarely – thrombocytopenia.
Immune system disorders:
Often – allergic reactions.
Very rarely – anaphylaxis.
Metabolic and nutritional disorders:
Often – hyperglycemia.
Uncommon: hypoglycemia, weight gain, anorexia. Unknown – diabetes mellitus: the incidence depends on the presence or absence of risk factors (fasting blood glucose concentration ≥ 5.6 mmol/l, body mass index (BMI) > 30 kg/m2, elevated triglyceride concentrations, history of arterial hypertension).
Mental disorders:
Infrequently – “nightmare” dreams, insomnia.
Unknown – depression.
Nervous system disorders:
Often – headache.
Uncommon: dizziness, paresthesia, hypoesthesia, impaired taste perception, amnesia.
Rarely – peripheral neuropathy.
Unknown – memory loss or decline.
Visual disorders:
Infrequently – the appearance of a “veil” before the eyes.
Rarely – visual impairment.
Hearing and labyrinthine disorders:
Uncommon: tinnitus.
Very rarely – hearing loss.
Disorders of the respiratory system, chest and mediastinal organs:
Often – sore throat, nosebleeds.
Unknown – isolated cases of interstitial lung disease (usually with long-term use).
Gastrointestinal disorders:
Often – constipation, flatulence, dyspepsia, nausea, diarrhea. Uncommon: vomiting, abdominal pain, belching, pancreatitis, abdominal discomfort. Liver and biliary tract disorders:
Uncommon – hepatitis.
Rarely – cholestasis.
Very rarely – renal failure.
Skin and subcutaneous tissue disorders:
Uncommon: urticaria, skin itching, skin rash, alopecia. Rarely – angioedema, bullous rash, polymorphic exudative erythema (including Stevens-Johnson syndrome), toxic epidermal necrolysis (Lyell’s syndrome).
Musculoskeletal and connective tissue disorders:
Often – myalgia, arthralgia, pain in the limbs, muscle cramps, swelling of the joints, back pain, musculoskeletal pain.
Uncommon: neck pain, muscle weakness.
Rarely – myopathy, myositis, rhabdomyolysis, tendinopathy (in some cases with tendon rupture).
Unknown – immune-mediated necrotizing myopathy.
Disorders of the genital organs and breast:
Infrequently – impotence.
Very rarely – gynecomastia.
General disorders and disorders at the injection site:
Uncommon: malaise, asthenic syndrome, chest pain, peripheral edema, fatigue, fever.
Influence on the results of laboratory and instrumental studies:
Often – deviation from the norm in the results of “liver” tests (AST and ALT), increased activity of serum creatine phosphokinase (CPK). Uncommon – leukocyturia.
Unknown – increased concentration of glycosylated hemoglobin (HbA1).
Children
Adverse reactions associated with taking Liprimar® did not differ in number from reactions caused by taking placebo. The most common reactions, regardless of control frequency, were infections.
Children and adolescents aged 10 to 17 years who were treated with atorvastatin had a side effect profile that was generally consistent with that of a patient receiving placebo. The most common adverse events observed regardless of causality assessment in both groups were infections. In a 3-year study assessing overall maturation and development, Tanner stage, and height and weight measurements, no clinically significant effects on height and puberty were observed. The safety and tolerability profile in children and adolescents was generally consistent with the known safety profile of atorvastatin in adults.
The clinical safety database also includes data from 520 pediatric patients treated with atorvastatin. Of these: 7 patients were under 6 years of age, 121 patients were between 6 and 9 years of age, and 392 patients were between 10 and 17 years of age. Based on available data, the frequency, type, and severity of adverse events in children are similar to those in adults.
The following undesirable side effects have been observed with the use of certain statins:
– sexual dysfunction;
– depression;
– in exceptional cases, especially with long-term therapy, interstitial lung disease (see section “Special instructions”);
– diabetes mellitus: frequency depends on the presence or absence of risk factors (fasting blood sugar ≥ 5.6 mmol/l, BMI > 30 kg/m2, elevated triglyceride levels, existing hypertension).
During treatment with HMG-CoA reductase inhibitors, with simultaneous use of cyclosporine, fibrates, nicotinic acid in lipid-lowering doses (more than 1 g / day) or inhibitors of the CYP3A4 isoenzyme/transport protein (for example, erythromycin, clarithromycin, antifungal agents – azole derivatives), the risk of myopathy increases (see sections “Dosage and Administration” “Special instructions”).
CYP3A4 isoenzyme inhibitors
Since atorvastatin is metabolized by the CYP3A4 isoenzyme, co-administration of atorvastatin with inhibitors of the CYP3A4 isoenzyme may lead to increased plasma concentrations of atorvastatin. The degree of interaction and potentiation effect is determined by the variability of the effect on the CYP3A4 isoenzyme.
It was found that potent inhibitors of the CYP3A4 isoenzyme lead to a significant increase in the concentration of atorvastatin in the blood plasma. The simultaneous use of strong inhibitors of the CYP3A4 isoenzyme (for example, cyclosporine, telithromycin, clarithromycin, delavirdine, stiripentol, ketoconazole, voriconazole, itraconazole, posaconazole and HIV protease inhibitors, including ritonavir, lopinavir, atazanavir, indinavir, darunavir, etc.) should be avoided whenever possible. If concomitant use of these drugs is necessary, initiating therapy at the lowest dose should be considered and the possibility of reducing the maximum dose of atorvastatin should be evaluated.
Moderate inhibitors of the CYP3A4 isoenzyme (for example, erythromycin, diltiazem, verapamil and fluconazole) may lead to increased plasma concentrations of atorvastatin. With the simultaneous use of HMG-CoA reductase inhibitors (statins) and erythromycin, an increased risk of developing myopathy was noted. Interaction studies between amiodarone or verapamil and atorvastatin have not been conducted. Both amiodarone and verapamil are known to inhibit the activity of the CYP3A4 isoenzyme, and concomitant use of these drugs with atorvastatin may lead to increased exposure to atorvastatin. In this regard, it is recommended to reduce the maximum dose of atorvastatin and carry out appropriate monitoring of the patient’s condition when used simultaneously with moderate inhibitors of the CYP3A4 isoenzyme. Monitoring should be carried out after the start of therapy and against the background of changing the dose of the inhibitor.
Gemfibrozil/fibrates
With the use of fibrates in monotherapy, adverse reactions, including rhabdomyolysis, affecting the musculoskeletal system were periodically noted. The risk of such reactions increases with simultaneous use of fibrates and atorvastatin. If the simultaneous use of these drugs cannot be avoided, the minimum effective dose of atorvastatin should be used, and the patient’s condition should be regularly monitored.
Ezetimibe
The use of ezetimibe is associated with the development of adverse reactions, including rhabdomyolysis, from the musculoskeletal system. The risk of such reactions increases with simultaneous use of ezetimibe and atorvastatin. For such patients, careful monitoring is recommended.
Erythromycin/clarithromycin
With the simultaneous use of atorvastatin and erythromycin (500 mg 4 times a day) or clarithromycin (500 mg 2 times a day), inhibitors of the CYP3A4 isoenzyme, an increase in the concentration of atorvastatin in the blood plasma was observed (see sections “Special instructions” and “Pharmacokinetics”).
Protease inhibitors
The simultaneous use of atorvastatin with protease inhibitors, known as inhibitors of the CYP3A4 isoenzyme, is accompanied by an increase in the concentration of atorvastatin in the blood plasma.
Diltiazem
The combined use of atorvastatin at a dose of 40 mg with diltiazem at a dose of 240 mg leads to an increase in the concentration of atorvastatin in the blood plasma (see section “Pharmacokinetics”).
Cimetidine
No clinically significant interaction of atorvastatin with cimetidine was detected (see section “Pharmacokinetics”).
Itraconazole
The simultaneous use of atorvastatin in doses from 20 mg to 40 mg and itraconazole in a dose of 200 mg led to an increase in the AUC value of atorvastatin (see section “Pharmacokinetics”).
Grapefruit juice
Since grapefruit juice contains one or more components that inhibit the CYP3A4 isoenzyme, its excessive consumption (more than 1.2 liters per day) may cause an increase in the concentration of atorvastatin in the blood plasma (see section “Pharmacokinetics”).
Transport protein inhibitors
Atorvastatin is a substrate of liver enzyme transporters (see section “Pharmacokinetics”).
Co-administration of atorvastatin 10 mg and cyclosporine 5.2 mg/kg/day resulted in increased atorvastatin exposure (AUC ratio: 8.7) (see Pharmacokinetics section). Cyclosporine is an inhibitor of organic anion transport polypeptide 1B1 (OATP1B1), OATP1B3, multidrug resistance-associated protein 1 (MDR1) and breast cancer resistance protein, as well as CYP3A4, and therefore increases the exposure of atorvastatin. The daily dose of atorvastatin should not exceed 10 mg (see section “Dosage and Administration”).
Glecaprevir and pibrentasvir are inhibitors of OATP1B1, OATP1B3, MDR1 and breast cancer resistance protein and therefore increase the exposure of atorvastatin. The daily dose of atorvastatin should not exceed 10 mg (see section “Dosage and Administration”).
Co-administration of atorvastatin 20 mg and letermovir 480 mg daily resulted in increased atorvastatin exposure (AUC ratio: 3.29) (see Pharmacokinetics section). Letermovir is an inhibitor of the transporters P-gp, BCRP, MRP2, OAT2 and the hepatic transporter OATP1B1/1B3, thus increasing the level of exposure to atorvastatin. The daily dose of atorvastatin should not exceed 20 mg (see section “Dosage and Administration”).
The magnitude of indirect drug interactions of CYP3A and OATP1B1/1B3 on co-administration of drugs may differ when letermovir is co-administered with cyclosporine. It is not recommended to use atorvastatin in patients receiving letermovir therapy in combination with cyclosporine.
Elbasvir and grazoprevir are inhibitors of OATP1B1, OATP1B3, MDR1 and breast cancer resistance protein and therefore increase the exposure of atorvastatin. Should be used with caution and at the lowest dose required (see section “Dosage and Administration”).
Inducers of the CYP3A4 isoenzyme
The combined use of atorvastatin with inducers of the CYP3A4 isoenzyme (for example, efavirenz, rifampicin or St. John’s wort preparations) may lead to a decrease in the concentration of atorvastatin in the blood plasma. Due to the dual mechanism of interaction with rifampicin (an inducer of the CYP3A4 isoenzyme and an inhibitor of the hepatocyte transport protein OATP1B1), simultaneous use of atorvastatin and rifampicin is recommended, since delayed administration of atorvastatin after taking rifampicin leads to a significant decrease in the concentration of atorvastatin in the blood plasma (see section “Pharmacokinetics”). However, the effect of rifampicin on the concentration of atorvastatin in hepatocytes is unknown and if concomitant use cannot be avoided, the effectiveness of this combination should be carefully monitored during therapy.
Antacids
Simultaneous oral administration of a suspension containing magnesium hydroxide and aluminum hydroxide reduced the concentration of atorvastatin in the blood plasma (change in AUC: 0.66), but the degree of reduction in the concentration of LDL-C did not change.
Phenazone
Atorvastatin does not affect the pharmacokinetics of phenazone, so interaction with other drugs metabolized by the same cytochrome isoenzymes is not expected.
Colestipol
With simultaneous use of colestipol, the concentration of atorvastatin in the blood plasma decreased (change in AUC: 0.74); however, the lipid-lowering effect of the combination of atorvastatin and colestipol was superior to that of each drug alone.
Digoxin
With repeated administration of digoxin and atorvastatin at a dose of 10 mg, the equilibrium concentrations of digoxin in the blood plasma did not change. However, when digoxin was used in combination with atorvastatin at a dose of 80 mg/day, digoxin concentrations increased (AUC change: 1.15). Patients receiving digoxin in combination with atorvastatin require appropriate monitoring.
Azithromycin
With simultaneous use of atorvastatin at a dose of 10 mg 1 time per day and azithromycin at a dose of 500 mg 1 time per day, the concentration of atorvastatin in the blood plasma did not change.
Oral contraceptives
With simultaneous use of atorvastatin and oral contraceptives containing norethisterone and ethinyl estradiol, increased concentrations of norethisterone (AUC change 1.28) and ethinyl estradiol (AUC change 1.19) were observed. This effect should be taken into account when choosing an oral contraceptive for a woman taking atorvastatin.
Terfenadine
With simultaneous use of atorvastatin and terfenadine, no clinically significant changes in the pharmacokinetics of terfenadine were detected.
Warfarin
In a clinical study in patients regularly receiving warfarin therapy, concomitant use of atorvastatin at a dose of 80 mg per day resulted in a slight increase in prothrombin time of approximately 1.7 s during the first 4 days of therapy. The indicator returned to normal within 15 days of atorvastatin therapy. Although significant interactions affecting anticoagulant function have been observed only in rare cases, the prothrombin time should be determined before initiating atorvastatin therapy in patients receiving coumarin anticoagulant therapy and frequently enough during therapy to prevent a significant change in the prothrombin time. Once stable prothrombin time values are observed, its monitoring can be carried out in the same way as recommended for patients receiving coumarin anticoagulants. When changing the dose of atorvastatin or discontinuing therapy, prothrombin time should be monitored according to the same principles as described above. Atorvastatin therapy was not associated with bleeding or changes in prothrombin time in patients not receiving anticoagulant treatment.
Colchicine
Although studies have not been conducted on the simultaneous use of colchicine and atorvastatin, there are reports of the development of myopathy when using this combination. Caution should be exercised when atorvastatin and colchicine are used concomitantly.
Amlodipine
In a drug interaction study in healthy subjects, coadministration of atorvastatin 80 mg and amlodipine 10 mg resulted in a clinically nonsignificant increase in atorvastatin concentrations (AUC change: 1.18)
Fusidic acid
During post-marketing studies, cases of rhabdomyolysis have been reported in patients taking concomitant statins, including atorvastatin and fusidic acid. The mechanism of this interaction is unknown. In patients for whom the use of fusidic acid is considered necessary, statin treatment should be discontinued for the entire period of use of fusidic acid. Statin therapy can be resumed 7 days after the last dose of fusidic acid. In exceptional cases where long-term systemic therapy with fusidic acid is necessary, for example for the treatment of severe infections, the need for co-administration of atorvastatin and fusidic acid should be considered on a case-by-case basis and under close medical supervision. The patient should seek immediate medical attention if symptoms of muscle weakness, tenderness, or pain occur.
Other concomitant therapy
In clinical studies, atorvastatin was used in combination with antihypertensive agents and estrogens as part of hormone replacement therapy. There were no signs of clinically significant adverse interactions; No interaction studies with specific drugs have been conducted.
In addition, an increase in the concentration of atorvastatin was observed when used simultaneously with HIV protease inhibitors (combinations of lopinavir and ritonavir, saquinavir and ritonavir, darunavir and ritonavir, fosamprenavir, fosamprenavir with ritonavir and nelfinavir), hepatitis C protease inhibitors (boceprevir, elbasvir/grazoprevir, simeprevir), clarithromycin and itraconazole. Caution should be exercised when using these drugs together and the lowest effective dose of atorvastatin should be used.
Effect of other drugs on the pharmacokinetics of atorvastatin
Cyclosporine 5.2 mg/kg/day, constant dose
Atorvastatin 10 mg once daily for 28 days
Change in AUC& – ↑ 8.7 times
Change in Cmax& – ↑ 10.7 times
Tipranavir 500 mg twice daily/ritonavir 200 mg twice daily for 7 days
Atorvastatin 10 mg, once
Change in AUC& – ↑ 9.4 times
Change in Cmax& – ↑ 8.6 times
Glecaprevir 400 mg once daily/pibrentasvir 120 mg once daily for 7 days
Atorvastatin 10 mg once daily for 7 days
Change in AUC& – ↑ 8.3 times
Change in Cmax& – ↑ by 22.00 times
Telaprevir 750 mg every 8 hours for 10 days
Atorvastatin 20 mg, once
Change in AUC& – ↑ by 7.9 times
Change in Cmax& – ↑ 10.6 times
Elbasvir 50 mg once daily/grazoprevir 200 mg once daily for 13 days
Atorvastatin 10 mg, once
Change in AUC& – ↑ by 1.95 times
Change in Cmax& – ↑ 4.3 times
Boceprevir 800 mg 3 times a day for 7 days
Atorvastatin 40 mg, once
Change in AUC& – ↑ by 2.3 times
Change in Cmax& – ↑ 2.7 times
Simeprevir at a dose of 150 mg 1 time per day for 10 days
Atorvastatin 40 mg, once
Change in AUC& – ↑ by 2.12 times
Change in Cmax& – ↑ 1.7 times
Lopinavir 400 mg twice daily/ritonavir 100 mg twice daily for 14 days
Atorvastatin 20 mg once daily for 4 days
Change in AUC& – ↑ by 5.9 times
Change in Cmax& – ↑ 4.7 times
‡ Saquinavir 400 mg twice daily/ritonavir 400 mg twice daily for 15 days
Atorvastatin 40 mg once daily for 4 days
Change in AUC& – ↑ by 3.9 times
Change in Cmax& – ↑ 5.4 times
Darunavir 300 mg twice daily/ritonavir 100 mg twice daily for 9 days
Atorvastatin 10 mg once daily for 4 days
Change in AUC& – ↑ by 3.4 times
Change in Cmax& – ↑ 2.2 times
Itraconazole 200 mg once daily for 4 days
Atorvastatin 40 mg, once
Change in AUC& – ↑ by 3.3 times
Change in Cmax& – ↑ by 1.20 times
Letermovir 480 mg once daily for 10 days
Atorvastatin 20 mg, once
Change in AUC& – ↑ by 3.29 times
Change in Cmax& – ↑ by 2.17 times
Fosamprenavir 700 mg twice daily/ritonavir 100 mg twice daily for 14 days
Atorvastatin 10 mg once daily for 4 days
Change in AUC& – ↑ 2.5 times
Change in Cmax& – ↑ 2.8 times
Fosamprenavir 1400 mg 2 times a day for 14 days
Atorvastatin 10 mg once daily for 4 days
Change in AUC& – ↑ by 2.3 times
Change in Cmax& – ↑ 4.0 times
Nelfinavir 1250 mg 2 times a day for 14 days
Atorvastatin 10 mg once daily for 28 days
Change in AUC& – ↑ by 1.74 times
Change in Cmax& – ↑ 2.2 times
Grapefruit juice, 240 ml once a day*
Atorvastatin 40 mg, once
Change in AUC& – ↑ by 1.37 times
Change in Cmax& – ↑ by 1.16 times
Diltiazem 240 mg once daily for 28 days
Atorvastatin 40 mg, once
Change in AUC& – ↑ by 1.51 times
Change in Cmax& – ↑ by 1.00 times
Erythromycin 500 mg 4 times a day for 7 days
Atorvastatin 10 mg, once
Change in AUC& – ↑ by 1.33 times
Change in Cmax& – ↑ by 1.38 times
Amlodipine 10 mg, once
Atorvastatin 80 mg, once
Change in AUC& – ↑ by 1.18 times
Change in Cmax& – ↑ by 0.91 times
Cimetidine 300 mg 4 times a day for 2 weeks
Atorvastatin 10 mg once daily for 2 weeks
Change in AUC& – ↓1.00 times
Change in Cmax& – ↓ 0.89 times
Colestipol 10 mg twice daily for 24 weeks
Atorvastatin 40 mg once daily for 8 weeks
AUC& change – Not established
Change in Cmax& – ↓ by 0.74** times
Maalox TC® 30 ml once a day, for 17 days
Atorvastatin 10 mg once daily for 15 days
Change in AUC& – ↓ 0.66 times
Change in Cmax& – ↓ 0.67 times
Efavirenz 600 mg once daily for 14 days
Atorvastatin 10 mg for 3 days
Change in AUC& – ↓ 0.59 times
Change in Cmax& – ↓ 1.01 times
Rifampicin 600 mg once daily for 7 days (concomitant use)†
Atorvastatin 40 mg, once
Change in AUC& – ↑ by 1.12 times
Change in Cmax& – ↑ by 2.9 times
Rifampicin 600 mg once daily for 5 days (separate doses)†
Atorvastatin 40 mg, once
Change in AUC& – ↓ 0.20 times
Change in Cmax& – ↓ 0.60 times
Gemfibrozil 600 mg 2 times a day for 7 days
Atorvastatin 40 mg, once
Change in AUC& – ↑ by 1.35 times
Change in Cmax& – ↓ 1.00 times
Fenofibrate 160 mg once daily for 7 days
Atorvastatin 40 mg, once
Change in AUC& – ↑ by 1.03 times
Change in Cmax& – ↑ by 1.02 times
& The relationship between the types of therapy is presented (co-administration of the drug with atorvastatin compared with the use of atorvastatin alone)
* With significant consumption of grapefruit juice (≥ 750 ml – 1.2 l per day), a greater increase in AUC (up to 2.5) and/or Cmax (up to 1.71) was noted;
** Based on a sample taken once 8-16 hours after taking the drug;
† Because rifampicin has a dual mechanism of interaction, it is recommended that atorvastatin and rifampicin be administered simultaneously. Later administration of atorvastatin after rifampicin is associated with a significant decrease in plasma concentrations of atorvastatin.
‡ The dosages of saquinavir/ritonavir used in this study differ from those used in clinical practice. It should be noted that the increase in atorvastatin exposure during clinical use is likely greater than that observed in this study. In this regard, the lowest dose of atorvastatin should be used.
Effect of atorvastatin on the pharmacokinetics of other drugs
Atorvastatin 80 mg once daily for 15 days
Antipyrine, 600 mg, once
Change in AUC& – ↑ by 1.03 times
Change in Cmax& – ↓ 0.89 times
Atorvastatin 80 mg once daily for 10 days
Digoxin 0.25 mg once daily for 20 days
Change in AUC& – ↑ by 1.15 times
Change in Cmax& – ↑ by 1.20 times
Atorvastatin 40 mg once daily for 22 days
Oral contraceptives once daily for 2 months: norethindrone 1 mg; ethinylestradiol 35 mcg
Change in AUC& – ↑ by 1.28 times
Change in Cmax& – ↑ by 1.23 times
Atorvastatin 10 mg, once
Tipranavir 500 mg twice daily/ritonavir 200 mg twice daily for 7 days
Change in AUC& – ↑ by 1.08 times
Change in Cmax& – ↑ by 0.96 times
Atorvastatin 10 mg once daily for 4 days
Fosamprenavir 1400 mg 2 times a day for 14 days
Change in AUC& – ↓ 0.73 times
Change in Cmax& – ↓ 0.82 times
Atorvastatin 10 mg once daily for 4 days
Fosamprenavir 700 mg twice daily/ritonavir 100 mg twice daily for 14 days
Change in AUC& – ↑ by 0.99 times
Change in Cmax& – ↑ by 0.94 times
& The relationship between the types of therapy is presented (co-administration of the drug with atorvastatin compared with the use of atorvastatin alone)
There is no specific antidote for the treatment of overdose with Liprimar®.
In case of overdose, symptomatic treatment should be provided as needed. Liver function tests should be performed and CPK activity should be monitored. Since the drug actively binds to plasma proteins, hemodialysis is ineffective.
Store at a temperature not exceeding 25 ºС.
Keep out of the reach of children.
3 years
Pfizer Pharmaceuticals LLC, Puerto Rico
| Shelf life | 3 years |
|---|---|
| Conditions of storage | At a temperature not exceeding 25 °C |
| Manufacturer | Pfizer Pharmaceuticals LLC, Puerto Rico |
| Medication form | pills |
| Brand | Pfizer Pharmaceuticals LLC |

Atherosclerosis
Atherosclerosis

Endocrinology
Atherosclerosis

Asthenia, weakness

Asthenia, weakness

Asthenia, weakness

Asthenia, weakness

Asthenia, weakness

Asthenia, weakness

Asthenia, weakness

Asthenia, weakness
Buy Liprimar, 20 mg 100 pcs with delivery to USA, UK, Europe and over 120 other countries.