-
×
 Ribavirin-SZ, 200 mg capsules 60 pcs
1 × €5.53
Ribavirin-SZ, 200 mg capsules 60 pcs
1 × €5.53 -
×
Enerion, 200 mg 20 pcs.
1 × €14.86
-
×
Liprimar, 20 mg 100 pcs
1 × €31.29
-
×
Crestor, 10 mg 126 pcs.
1 × €122.81
-
×
Eleutherococcus extract liquid, 50 ml
1 × €3.23
-
×
L-Carnitine Rompharm, 200 mg/ml 5 ml 5 pcs
1 × €13.99
-
×
Carniton, tablets, 20 pcs.
1 × €13.09
-
×
 Diroton, tablets 5 mg, 56 pcs.
1 × €5.91
Diroton, tablets 5 mg, 56 pcs.
1 × €5.91 -
×
Elcar, 300 mg/ml 100 ml
1 × €19.26
 Ribavirin-SZ, 200 mg capsules 60 pcs
Ribavirin-SZ, 200 mg capsules 60 pcs  Enerion, 200 mg 20 pcs.
Enerion, 200 mg 20 pcs.  Liprimar, 20 mg 100 pcs
Liprimar, 20 mg 100 pcs  Crestor, 10 mg 126 pcs.
Crestor, 10 mg 126 pcs.  Eleutherococcus extract liquid, 50 ml
Eleutherococcus extract liquid, 50 ml  L-Carnitine Rompharm, 200 mg/ml 5 ml 5 pcs
L-Carnitine Rompharm, 200 mg/ml 5 ml 5 pcs  Carniton, tablets, 20 pcs.
Carniton, tablets, 20 pcs. 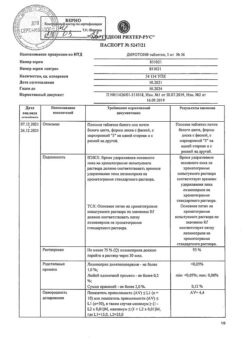 Diroton, tablets 5 mg, 56 pcs.
Diroton, tablets 5 mg, 56 pcs.  Elcar, 300 mg/ml 100 ml
Elcar, 300 mg/ml 100 ml Subtotal: €229.97